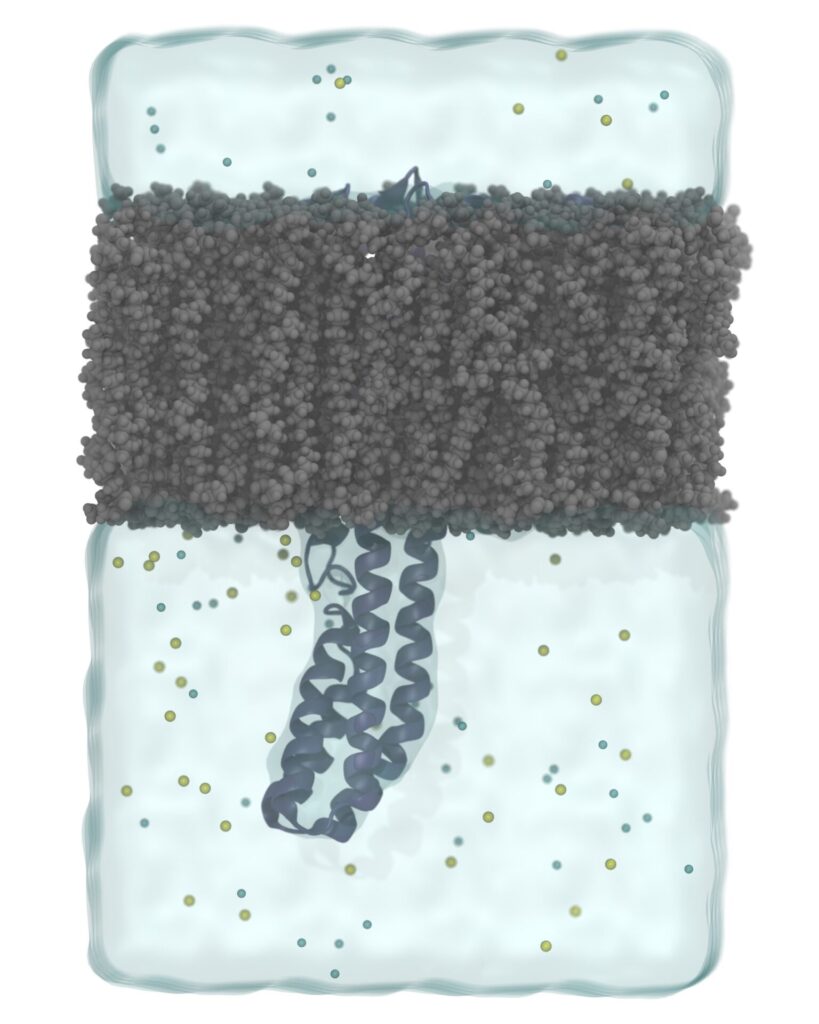
MD simulations describe the movement, interactions, and dynamics of biomolecules (e.g., proteins and nucleic acids) at the atomic level. MD simulations employ a “force field” based methodology that reflect all interatomic interactions and analyze the time dependent behaviour of biomolecules using Newton’s laws of motion. Combining MD with other CADD based techniques allows researchers to have a more realistic approximation of the biological systems. Our molecular dynamics simulation services include conventional long-run (> 1 µs) all-atom and coarse-grained MD simulations. For specific applications, we further employ enhanced-sampling techniques such steered molecular dynamics, adaptive biasing force, replica exchange MD, gaussian-accelerated MD and constant-pH MD simulations. In addition, we run allostery and druggability simulations to predict druggable binding sites of various protein targets by employing long-run mixed-solvent MD simulations.