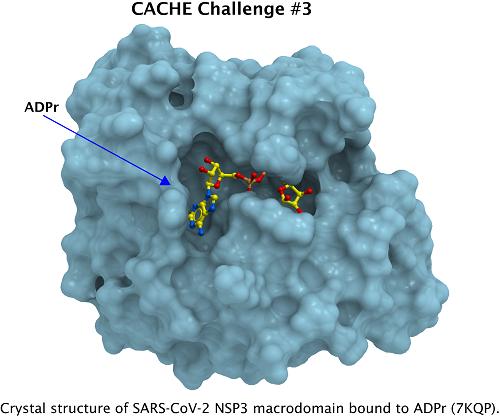
Dr. Sharma is a Computational Sciences specialist at Binary Star. He has more than fifteen years of experience across the breadth of computational sciences, including, Computational Chemistry, Computational Biochemistry, and Computer aided molecular and drug design. Previously, he served as a Principal Investigator of the Computational Biochemistry Laboratory, Department of Chemistry, Panjab University, Chandigarh, India, where he led a team conducting research in computational drug discovery, nucleic acids structural bioinformatics and DNA damage and repair processes. Prior to that, he was a INSPIRE faculty at Central University of Punjab, India.
While at Binary Star, he contributes to development and application of CADD protocols for GPCRs based drug design and focuses on different aspects of nucleic acid-based therapeutics. In addition, he works on the applications of AI and machine learning in solving big data-based problems related to health and informatics. Dr. Sharma received his Ph.D. in Computational Natural Sciences from International Institute of Information Technology, Hyderabad in India. He also completed a Postdoctoral Fellowship in Computational Chemistry at the Department of Chemistry and Biochemistry, University of Lethbridge, Alberta, Canada, where he focused on the role of nucleic acids in diseases and disorders.