Exploring the Role of Zinc in Alzheimer’s Disease: Insights from Metal Chemistry and Simulations
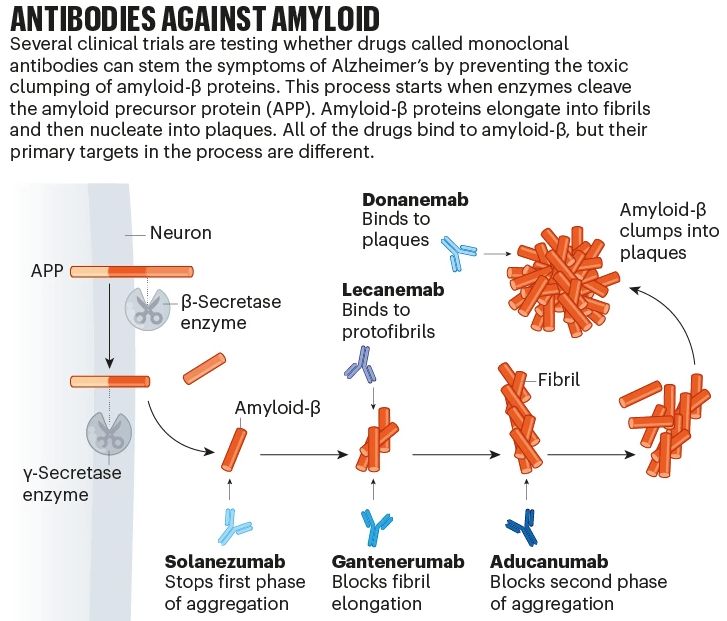
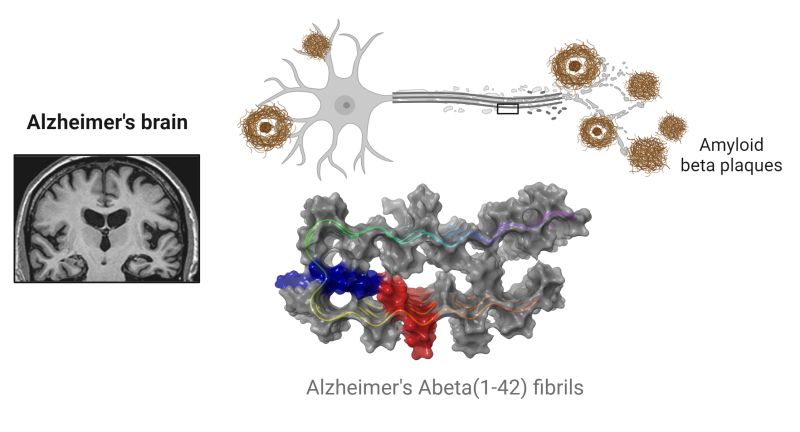
Metal Chemistry of Alzheimer’s Disease The metal ion hypothesis proposed that AD was associated with dyshomeostasis of metal ions, most particularly Fe, and later Zn and Cu. Significant advancements at the interface of bioinorganic chemistry and neurology have paved the path for intriguing aims related to zinc homeostasis. While the concentration of Zn2+ in aggregates is a key factor in Alzheimer\’s disease, the mechanism by which Zn2+/Aβ enhances aggregation is still unknown. By using MD and REMD simulations, Zinc coordination in A oligomers was discovered to reduce oligomer solvation and so promote Zn2+/ Aβ aggregation, resulting in less uniform structures. simulations revealed Zn2+ ions can bind at various sites in the N-terminal domain, resulting in structural variance with a number of populations. https://lnkd.in/gPm93kBn